– описание материалов, вступающих в непосредственный или опосредованный контакт с телом человека;
– перечень основных характеристик, размеров и указаний по эксплуатации медицинского изделия, его исполнений и принадлежностей, которые имеются в технической документации медицинского изделия и других материалах, доступных конечному пользователю, а также перечень применяемых производителем стандартов;
– информацию, позволяющую получить общее представление об основных стадиях проектирования рассматриваемого медицинского изделия. Данная информация может быть представлена в виде блок- схемы процессов;
– информацию, позволяющую получить общее представление о производственных процессах. Информация может быть представлена в виде блок-схемы процессов, дающей общее представление о производстве, сборке, заключительных испытаниях медицинского изделия и окончательной упаковке готового медицинского изделия;
– идентификацию производственных площадок, на которых осуществляется производственная деятельность по рассматриваемому медицинскому изделию;
– сведения о биологической совместимости;
– сведения о лекарственных средствах, входящих в состав рассматриваемого медицинского изделия;
– сведения о биологической безопасности медицинских изделий, включающих в себя клетки, ткани или их производные, взятые у человека или животных;
– сведения о методах стерилизации.
Основные требования к медицинским изделиям для диагностики in vitro включают:
– назначение изделия, в том числе (если применимо):
– описание целевого аналита, включая указание на качественный, полуколичественный или количественный вид определения;
– функциональное назначение;
– специфическую патологию, состояние или фактор риска, для обнаружения, определения или дифференцирования которого предназначено изделие;
– тип анализируемого образца;
– потенциальных пользователей;
– описание принципа аналитического метода или принципа действия прибора;
– описание составных частей, в том числе перечень возможных вариантов исполнения рассматриваемого изделия;
– описание принадлежностей, других изделий (в том числе медицинских), которые предполагается использовать в сочетании с изделием;
– описание материалов для взятия и транспортировки образцов или описание (характеристики) материалов, рекомендуемых для этой цели;
– технические характеристики (для аналитического оборудования);
– информацию об основных стадиях проектирования рассматриваемого медицинского изделия для диагностики in vitro. Данная информация может быть представлена в виде блок-схемы процессов;
– информацию о производственных процессах, в том числе о производстве, сборке, заключительных испытаниях медицинского изделия для диагностики in vitro и окончательной упаковке готового медицинского изделия для диагностики in vitro;
– сведения о производственных площадках, на которых осуществляется производство рассматриваемого медицинского изделия для диагностики in vitro.;
– сведения об аналитической эффективности медицинского изделия для диагностики in vitro (если применимо):
– аналитическую чувствительность (порог обнаружения);
– аналитическую специфичность;
– правильность определений;
– случайную ошибку;
– данные о метрологической прослеживаемости значений калибраторов и контрольных материалов;
– данные об аналитическом диапазоне (диапазоне линейности – для линейных аналитических систем), а также описание методов определения характеристик;
– данные об определении «точки отсечки» (cut-off), включая описание деталей метода определения характеристик;
– популяционные (демографические) аспекты применения медицинского изделия для диагностики in vitro.
6.3.7 Понимание применимого законодательства и стандартов
Для медицинских изделий в зависимости от стратегии регистрации могут быть применимы национальное законодательство РФ или ЕАЭС. Необходимо установить, какие правила и стандарты могут повлиять на конструкцию. В некоторых случаях подход к проектированию может диктовать, применимы ли конкретные стандарты.
Пример:
В Европе изделие с питанием от сети обычно подпадает под действие стандарта по низкому напряжению (LV) (требуется технический файл и маркировка CE), тогда как такое же изделие, работающее от сетевого адаптера, не подпадает под действие Директивы по низковольтному оборудованию (хотя сам адаптер должен). Таким образом, использование последнего подхода может значительно ослабить ограничения на проектирование и производство изделия.
Спецификация должна включать ссылки на любые стандарты, которым должно соответствовать изделие (например, ГОСТ). Если эти стандарты, в свою очередь, налагают особые требования, которые еще не заложены в конструкцию, то эти требования должны быть перечислены в спецификации (например, требование к материалу печатной платы с ограниченной воспламеняемостью, стандарт UL 94v0, в электронном изделии). Если стандарты устанавливают требования, которые будут выполняться по умолчанию, то эти требования также должны быть перечислены в спецификации.
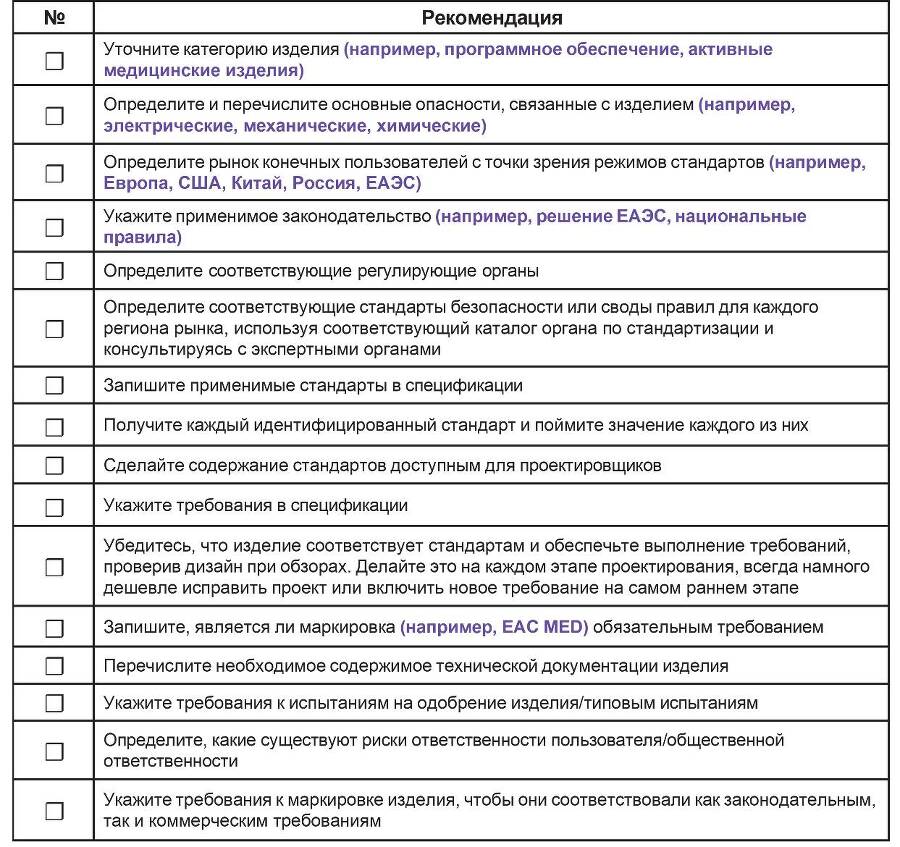
В таблице 6.4 приведен рекомендуемый список для определения применимого законодательства и стандартов.
Таблица 6.4 – Контрольный список рекомендаций для определения применимого законодательства и стандартов
6.3.8 Понимание того, как производить изделие
После того, как появится детальный проект (этап реализации), параметры изделия могут быть окончательно оформлены в спецификации. Архитектура изделия и рабочий проект по его созданию теперь могут быть полностью зафиксированы в проектной документации.
Критерии производства должны быть установлены как можно более подробно, включая методы производства и испытаний. В этом должен участвовать производственный персонал (включая сотрудников любых крупных субподрядчиков). Любые специальные критерии для покупных деталей или узлов должны быть зарегистрированы.
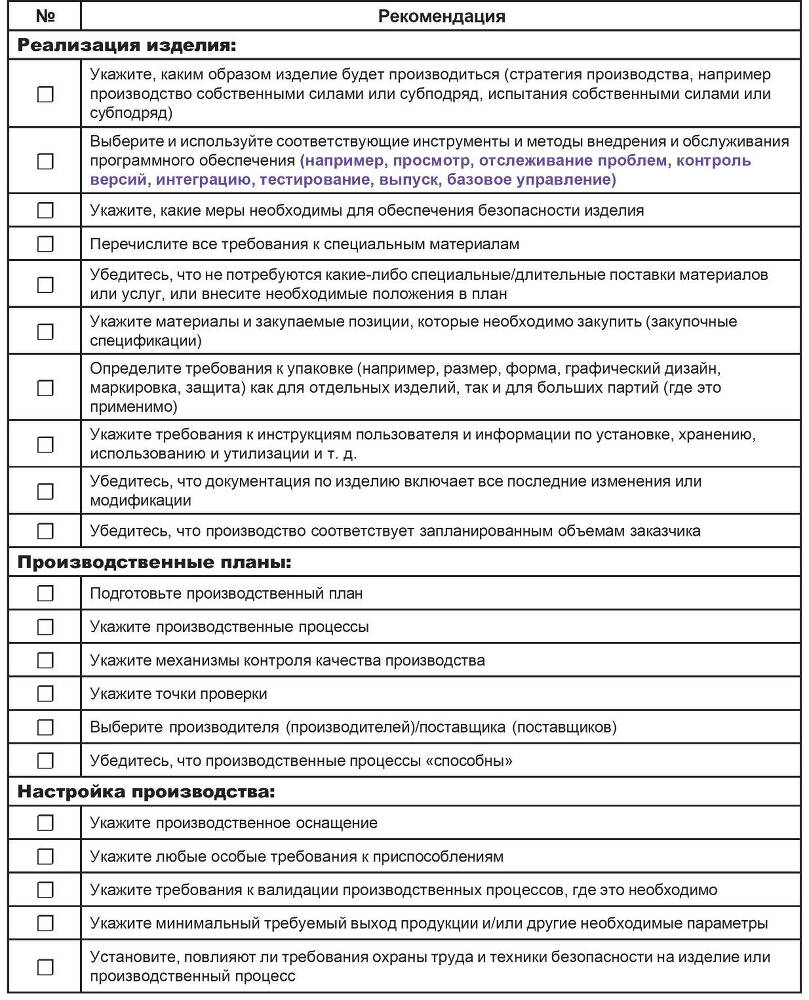
В таблице 6.5 приведен рекомендуемый список для использования при рассмотрении вопроса о том, как производить изделие в общем случае.
Таблица 6.5 – Контрольный список рекомендаций для использования при рассмотрении вопроса о том, как производить изделие
6.3.9 Понимание того, как верифицировать, тестировать и валидировать изделие
При установлении процесса соответствия изделия важно различать верификацию, производственное испытание и валидацию.
– Верификация устанавливает, что изделие соответствует его спецификации. В какой-то степени это может быть достигнуто в процессе проектирования с помощью анализа проекта и т. д. Затем это подтверждается окончательной записью, показывающей, что изделие соответствует всем требованиям.
– Производственные испытания гарантируют, что отдельные образцы изделия функционируют удовлетворительно.
– Валидация в своей простейшей форме устанавливает, что изделие полностью соответствует своему назначению, то есть изделие успешно функционирует в клинической среде.
В процессе верификации и валидации следует учитывать все атрибуты изделия. Производственные испытания охватывают критические аспекты и/или аспекты, связанные с безопасностью, и являются лишь подмножеством общих характеристик изделия. Обычно необходимо полностью протестировать образцы изделия, чтобы получить данные, необходимые для первоначальной проверки проекта. Валидация будет включать клинические испытания изделия в рабочих условиях, чтобы убедиться, что он соответствует потребностям клиента. Валидация также будет включать запись данных в качестве доказательства процесса валидации. Для некоторых изделий валидация может быть достижима только при тестировании в предназначенной среде использования.