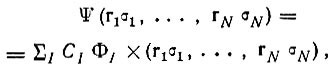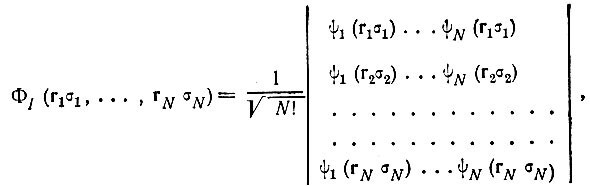Исследования "химико-алгебраической аналогии", начатые Сильвестром и Клиффордом, были затем продолжены в работах по теории инвариантов В. Г. Алексеева [1, 45]. Различаясь по способу математической интерпретации постулатов теории химического строения, эти работы оказались сходными в трактовках понятий валентности и химической связи, которые обладали следующими особенностями: а) валентность того или иного элемента определялась из химических, а не из математических соображений; б) валентность явно или неявно выступала как сумма кратностей связей, образуемых данным атомом с другими, причем единичной химической связи между атомами X и Y соответствовал одночленный инвариант [XY].
Указанная аналогия была скептически встречена некоторыми математиками (Нетер, Штуди). Штуди, например, писал, что "было бы слишком фантастическим ожидать, будто химия когда-либо извлечет пользу из этой ветви алгебры". Однако появлением квантовой механики положение изменилось. Если в XIX — начале XX вв. речь шла, по словам Вейля, "о исто формальной, хотя и очень впечатляющей математической аналогии", то в конце 20-х годов говорится уже о "существеннейшем звене в квантовомеханической теории химической связи, которой указанная аналогия имеет ... удивительно конкретное воплощение" (Вейль).
Выше мы рассмотрели основные направления теоретических исследований, которые подготовили почву для перехода от классической теории химического строения к квантовомеханической. Итак, можно выделить два подготовительных этапа создания квантовой теории химической связи. Первый, формально-математический, включает работы 1870 г.- 1900-х гг. (Кэли, Сильвестр, Клиффорд, Гордан, В. Г. Алексеев), в которых были предприняты попытки построения математических моделей молекул, основанные на теории инвариантов бинарных форм и теории химического строения. Второй подготовительный этап, электронный (1900-1926 гг.), включает работы, посвященные созданию электронных (как статических, так и динамических) моделей атомов и молекул.
При этом на обоих подготовительных этапах каждой ковалентной связи (валентному штриху) сопоставлялись некоторые дискретные объекты: либо это были компоненты двумерных векторов в формально-математических моделях, либо — отдельные электроны, занимающие определенные положения в атомах и молекулах (статические модели) или же двигающиеся по определенным траекториям (динамические модели). Как в математических, так и в физико-химических работах содержались рациональные идеи, вошедшие после соответствующей кванто-вомеханической интерпретации в квантовую химию: идея двухцентровой двухэлектронной связи (Льюис), возможность обобществления нескольких электронов вокруг двух и большего числа ядер (Ленгмюр), идея неподеленной электронной пары (Льюис, Сиджвик), разделение электронных и ядерных движений (Вор, Ван Флек, Зоммерфельд и др.), идея одноэлектронного приближения (Бор), сопоставление ковалентной связи некоторого математического выражения [XY], антисимметричного относительно перестановки своих компонент (Сильвестр, Алексеев и др.) и т. п.
Рис. 3. Линейно-независимые инварианты и соответствующие им структуры бензола
Создание в 1925-1926 гг. квантовой механики позволило глубоко проникнуть в сущность явлений и процессов, протекающих в атомах и молекулах, выявить физический смысл понятия химической связи и других понятий классической химии.
В конце 20-х годов были установлены общие квантовомеханические принципы и приближения, необходимые для описания многоэлектронных систем. Как и в период разработки электронных моделей (1900-1926 гг.) строения вещества, каждая квантовомеханическая модель химической связи существенно опиралась на квантовомеханическую (шредингеровскую) модель атома. На протяжении всей последующей эволюции теории многоэлектронных систем указанная взаимосвязь между теорией атома и теорией молекул сохранялась. Поэтому прежде чем приступить к рассмотрению основных этапов развития молекулярной квантовой химии и некоторых ее современных проблем, в следующей главе мы остановимся на основных понятиях и представлениях квантовой теории строения атома.
Глава 2. Квантовомеханическое описание строений атома
В настоящей главе кратко изложены некоторые результаты квантовомеханической теории строения атома, причем основное внимание уделено тем ее аспектам, которые представляют интерес для теории химической связи.
Некоторые особенности квантовомеханического описания явлений микромира
В квантовой механике состояние N-электронной системы описывается волновой функцией
зависящей от пространственных координат (r
i) и спиновых переменных (σ
i) всех электронов. Эта функция должна удовлетворять уравнению Шредингера
(2.1)
где
— оператор Гамильтона (гамильтониан), определяющий рассматриваемую систему и для атома с зарядом ядра Z,имеющий вид
(2.2)
Не следует думать, что любое решение уравнения Шредингера (2.1) имеет физический смысл. В действительности на функцию Ψ накладываются определенные ограничения. В частности, для связанных состояний с дискретным спектром Е должно выполняться условие нормировки:
(2.3)
Для многоэлектронных систем чрезвычайно важным является требование антисимметричности волновой функции
относительно перестановок тождественных частиц (электронов):
(2.4)
Одним из следствий этого требования, называемого принципом Паули, является то, что для трех и большего числа электронов основным состоянием не будет состояние с наименьшим собственным значением гамильтониана, так как последнему соответствует не имеющая физического смысла симметричная волновая функция.
Значение принципа Паули в теории химической связи исключительно велико, и мы в дальнейшем неоднократно будем к нему обращаться. Сейчас отметим только, что любую антисимметричную N-электронную функцию Ψ можно представить в виде линейной комбинации так называемых "детерминантов Слэтера", составленных из одноэлектронных волновых функций (спин-орбиталей):
(2.5)
где
(2.6)
и индекс I при детерминанте Слэтера ΦI обозначает определенную совокупность ортонормированных спин-орбиталей ψi(rσ), которая называется спин-орбитальной конфигурацией.
В общем случае в разложение многоэлектронной волновой функции входит бесконечно много детерминантов Слэтера. Часто ограничивают это разложение одним или несколькими детерминантами. Качество такой аппроксимации зависит от качества включенных в ΦI спин-орбиталей. Наилучшие спин-орбитали получаются в методе самосогласованного поля Хартри-Фока, на котором мы подробно остановимся в третьей главе.
Электронные конфигурации атомов, термы и тонкая структура энергетических уровней
Атомные спин-орбитали, описывающие одноэлектронные состояния в атоме, приближенно (без учета спин-орбитального взаимодействия) можно представить в виде произведения бесспиновой одноэлектронной волновой функции, называемой орбиталью, на одноэлектронную спиновую функцию, которая является собственной функцией оператора проекции собственного момента импульса электрона