A B C D
A .6 .6 .2
B .4 .6
C .6
а. Используя любой подход, который пожелаете, определите правильное некорневое дерево, относящееся к этим таксонам, а также все длины его ребер. Объясните, как исключили другие топологические деревья.
б. Можете ли определить корень дерева по этим данным? Объясните, почему да или почему нет.
Примечание: Методы решения такого рода проблем являются предметом следующих разделов.
5.2. Построение дерева дистанционными методами UPGMA и FM
При построении филогенетического дерева таксоны, которые хотим связать, обычно являются теми, которые живут в настоящее время. Есть информация, такая как последовательности ДНК, от терминальных таксонов и нет информации от тех, которые представлены внутренними вершинами. В действительности, даже не знаем, какие внутренние вершины должны существовать, потому что не знаем даже топологию дерева.
Первым классом методов построения филогенетических деревьев, которые обсудим, являются дистанционные методы. Они пытаются построить дерево, используя информацию, которая предположительно описывает общие расстояния между терминальными таксонами вдоль дерева.
Чтобы понять, как получить эти расстояния, представьте, что пытаемся найти эволюционные отношения четырех видов:
,
,
и
. Выбирая тот или иной ортологичный участок ДНК из их геномов, получаем и выравниваем последовательности из каждого. Если модель замены оснований Джукса-Кантора, рассмотренная в главе 4, кажется подходящей для имеющихся данных, то вычисляем расстояния Джукса-Кантора между каждой парой последовательностей. Получатся оценки расстояний по дереву, которые сводим в Таблицу 5.2.
В зависимости от данных последовательности могли бы вместо этого принять другую модель подстановки оснований, что привело бы к использованию другой формулы расстояния, такой как в 2-параметрической модели Кимуры или логарифмическое расстояние. Несмотря на это, расстояние, которое вычисляем между последовательностями, считается мерой количества произошедших мутаций. Если бы эти расстояния были точной мерой количества произведенных мутаций, они бы соответствовали между конечными таксонами в найденном метрическом дереве.
Таблица 5.2. Расстояния между таксонами
.45 .27 .53
.40 .50
.62
На самом деле даже не ожидаем найти дерево, которое точно соответствует имеющимся данным; в конце концов, расстояния выводятся из данных последовательности и не должны быть точно правильными. Более того, метод вывода расстояний зависел от модели, которая включала дополнительные предположения, которые, безусловно, не встречаются в реальных организмах. Надеемся, однако, что построенное дерево не будет слишком чувствительно к такого рода ошибкам на больших расстояниях.
Первый метод, который рассматриваем, называется методом среднего расстояния или, более формально, невзвешенным парно-групповым методом с арифметическими средними (UPGMA). Этот метод создает корневое дерево и предполагает наличие молекулярных часов. Самый простой способ понять алгоритм – это ознакомиться с примером его использования.
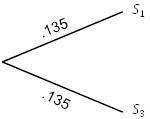
По приведенной выше таблицы данных выберем два ближайших таксона,
и
. Поскольку они находятся на расстоянии 0,27 друг от друга, изобразим на рисунке 5.6 каждое ребро с длиной
.
Рисунок 5.6. UPGMA; шаг 1.
Затем объединяем
и
в группу и усредняем расстояния
и
до каждого отдельного таксона, чтобы получить расстояние от группы до этого таксона. Например, расстояние между группой
и
равно
, а расстояние между
и
равно
. Таким образом, исходная таблица сводится к таблице 5.3.
Таблица 5.3. Расстояния между групп; UPGMA, Шаг 1
.425 .575
.50
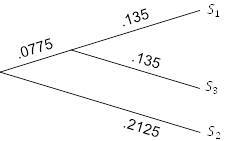
Теперь просто повторяем процесс, используя расстояния в таблице 5.3. Поскольку ближайшими таксонами и/или группами в новой таблице являются
и
, которые находятся на расстоянии 0,425 друг от друга, то получаем рисунок 5.7.
Рисунок 5.7. UPGMA; шаг 2.
Ребро
должно иметь длину
, в то время как другое новое ребро должно иметь длину
, потому что уже есть ребро длины
для учета некоторого расстояния между
и другими таксонами.