По внешнему виду нельзя отличить гетерозиготу по аутосомно-рецессивному гену от нормального индивида, Однако на биохимическом уровне летальный ген будучи в гетерозиготном состоянии проявляет свое действие. Если у гомозигот (аа) аутосомно-рецессивный ген обусловливает полное отсутствие активности какого-либо фермента и у индивида с таким генотипом диагностируется наследственное заболевание, то у гетерозигот (Аа) активность этого фермента не достигает 100 % (как у нормальных индивидов (Аа)), однако и наследственное заболевание у них не диагностируется. При тщательном клиническом обследовании у гетерозигот можно обнаружить небольшие отклонения от нормы.
Описано много наследственных болезней с аутосомно-рецессивным характером (типом) наследования, однако далеко не для всех из них известно место дефекта в цепи химических превращений, протекающих в клетке.
Среди аутосомно-рецессивных заболеваний самыми распространенными являются фенилкетонурия, галактоземия, цистинурия, гистидинемия, болезнь Тей-Сакса, мукополисахаридозы, муковисцидоз и др. Наиболее хорошо изучены фенилкетонурия (фенилпиро-виноградная олигофрения) и галактоземия (табл. 8).
Впервые фенилкетонурия описана в 1934 году ученым-генетиком Феллингом. Эта болезнь характеризуется тяжелыми поражениями нервной системы и выражается в умственной отсталости. По данным разных авторов, во всех странах мира до 3 % населения являются умственно неполноценными. Известно несколько сот наследственных болезней, сопровождающихся умственной отсталостью или ведущих к ней. Особенность фенилкетонурии в том, что она проявляется на первом году жизни ребенка и медленно прогрессирует. Без рано начатого лечения это заболевание делает ребенка инвалидом на всю жизнь, так как вызывает отравление клеток мозга (интоксикация мозга). Раннее выявление фенилкетонурии возможно только методом массового скринирования (обследования) новорожденных.
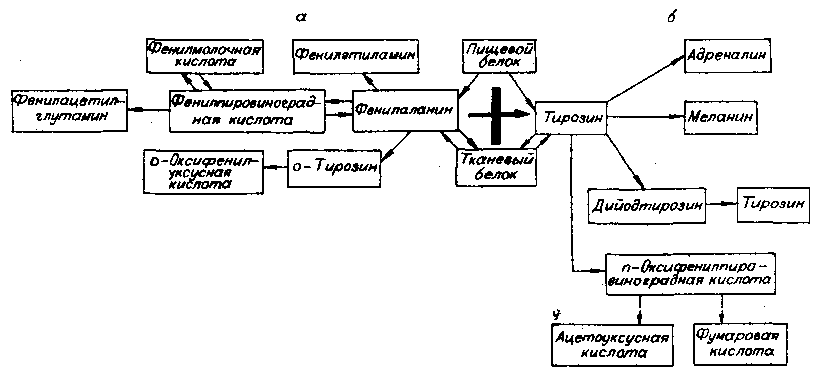
В нашей стране массовый скрининг новорожденных проводился впервые в 1975 году К. Д. Краснопольской в Москве и Ленинграде. Для фенилкетонурии известно, что аминокислота фенилаланин, поступающая в организм с молоком матери или детским питанием, не превращается в тирозин (следующую аминокислоту в цепи аминокислотного обмена) из-за недостаточной активности фермента фенилаланингидроксилазы (всего от 0 до 5 % активности нормы) (рис. 20). Частота, встречаемости фенилкетонурии оказалась высокой — 1 : 10 000 новорожденных, хотя в относительных цифрах и уступала таковой ряда стран (см. табл. 8).
Не останавливаясь более подробно на этом заболевании, отметим, лишь, что вскармливанием детей на специальной диете можно предотвратить интоксикацию мозга и последующую инвалидность и вырастить нормального гражданина общества. Очень важно, чтобы лечение было начато в первые два месяца жизни (и чем раньше, тем лучше). Только в этом случае оно будет эффективным. С чем это связано? М. Д. Армстронг и другие ученые-генетики показали, что у детей, больных фенилкетонурией, количество фенилаланина в крови изменяется с возрастом — а именно: у Детей до трех лет его примерно в 2 раза выше, чем у детей более старшего возраста. Выделение же фенилпировиноградной кислоты, отравляющей мозг, прямо коррелирует о количеством фенилаланина. Иными словами, в период наибольшего насыщения крови фенилаланином (в возрасте двух-трех лет) происходит наибольшая интоксикация мозга, и именно в этом возрасте ребенок наиболее остро нуждается в лечении.
«Лечение» фенилкетонурии начало проводиться в ряде стран с 1957—1958 годов. К настоящему времени из этих леченых детей выросли уже женихи и невесты. Однако слово «лечение» взято в кавычки не случайно, потому что дефект гена не исчез, не исправлен, он остался, но больному создана такая пищевая среда, что гену нет возможности проявиться, реализоваться, то есть, как говорят генетики, выйти на фенотип.
Рис. 20. Локализация генетического блока при фенилкетонурии. а, б — путь метаболизма фенилаланина соответственно у больных и здоровых.
Рассмотрим случай: жених здоров, у него нет слабоумия, однако гены в обеих матрицах остались гомозиготными — аа. Он собирается организовать семью. Если окажется, что у невесты нет ни одного гена, обусловливающего появление фенилкетонурии, то все их дети будут гетерозиготы (Аа) — то есть здоровы. Если же он полюбит девушку, гетерозиготную по гену фенилкетонурии (Аа), то в 50 % случаев можно ожидать рождения больного ребенка.
Теперь рассмотрим случай, когда «вылеченная» девушка стала невестой и собирается выйти замуж. Будет ли картина аналогичной предыдущей? Здесь все оказалось сложнее. «Вылеченная» девушка собирается стать матерью. Генетический дефект же у нее остался. Аминокислота фенилаланин в повышенной концентрации уже не может повлиять на ее мозговые клетки, однако будет губительно действовать на мозговое вещество ребенка, находящегося в утробном развитии.
Каролин и Стивенсон в 1969 году описали течение беременности у двух женщин, больных фенилкетонурией. 26 беременностей у пациенток закончились 16 спонтанными абортами в первые три месяца и 10 живорожденными детьми. Уровень фенилаланина в крови детей был выше, чем в крови матери. Живорожденные дети имели следующие пороки развития: микроцефалию, врожденные пороки сердца, умственную отсталость и др.
По логике, фенилаланин должен быть в повышенных концентрациях и у женщин, гетерозиготных из-за недостаточной активности фермента. Медики-генетики стали искать женщин-гетерозигот по фенилкетонурии среди населения и наблюдать за течением беременностей у них. Оказалось, что только у 6 из 46 таких женщин беременность и роды протекали нормально.
Из сказанного ясно, что гетерозиготное носительство гена фенилкетонурии со стороны матери, не говоря уже о гомозиготном (больная мать),— чрезвычайно неблагоприятное явление для беременности и родов.
Несколько слов о гениальности и таланте. Гениальность, как и талант человека, является результатом взаимодействия генотипа (со всем комплексом его врожденных данных) и окружающей среды. Соотношение этих двух слагаемых для каждого конкретного признака (свойства) разное, однако оба они в развитии вышеуказанных качеств необходимы. При благоприятной среде и отсутствии необходимого комплекса генов эти качества не разовьются. И, напротив, при предрасположенности к тому или иному дарованию, не подкрепленной необходимой социальной средой, результат будет аналогичным. Однако, опираясь на данные современной науки, можно констатировать, что в гениальности ученого, поэта, писателя, художника и т. д. вклад генотипа преобладает.
Но здесь нет места для пессимизма. Каждый здоровый человек талантлив по-своему и представляет для общества громадную ценность, и задача общества (и семьи) в том, чтобы это качество выявить и развить.
Современная генетика доказала, что человек является продуктом биосоциальным, а не «чисто социальным», как утверждали классики, и что его биологическая компонента составляет около 70%, а такие властные инстинкты, как размножение, самосохранение, забота о потомстве и т. д.,— все 100 %.
Гены, сцепленные с половыми хромосомами
К настоящему времени известно более 200 генов (доминантных и рецессивных), сцепленных с Х-хромосомой, то есть локализованных в ней. Многие из них обусловливают появление наследственных болезней (гемофилия, мышечная дистрофия и др.) (рис. 21).
У женщин аномальный ген может находиться в одной (гетерозигота) или обеих (гомозигота) Х-хромосомах, у мужчин — только в одной Х-хромосоме. Наиболее широко распространено заболевание, обусловленное геном, сцепленным с Х-хромосомой и имеющим рецессивный характер наследования,— гемофилия (несвертываемость крови) (рис. 22). В данном случае ген имеет полулетальное значение, что, как правило, приводит к гибели ребенка в раннем возрасте. Долгое время считалось, что этим заболеванием страдают только мужчины. Однако оно встречается и у женщин, хотя и крайне редко, и обусловлено наличием аномальных генов в обеих Х-хромосомах.