Для анализа распределения ЛС в органах и тканях используется понятие «кажущийся объем распределения» – это показатель, который направлен на то, чтобы предсказать, насколько равномерно лекарство распределяется по организму. Кажущийся объем распределения, Vd, рассчитывают путем деления введенной дозы (мг) на концентрацию в плазме (мг/л).
Vd (л) = доза (мг) / C (мг/л)
Показатель Vd равен условному объему (компартменту), в котором ЛС должно распределиться, чтобы достичь существующей концентрации С в плазме крови. Лекарства с относительно небольшим Vd (например, 5 л) в основном остаются в плазме крови. Лекарства с Vd = 15 л распределяются по сосудистым и внеклеточным жидким компартментам. Такой показатель Vd характерен для небольших гидрофильных молекул, которые покидают сосудистое русло благодаря парацеллюлярному транспорту, но не могут попасть внутрь клеток через липидные мембраны их оболочек. Препараты с Vd>40 л обладают высокой липофильностью и распределяются по всем тканям организма (сосудистым, внеклеточным и внутриклеточным жидкостным компартментам).
На кажущийся объем распределения оказывает влияние связывание с белками плазмы и депонирование. Так, липофильные вещества (например, неингаляционное средство для наркоза тиопентал) перераспределяются из плазмы в жировую ткань и, накапливаясь там, значительно увеличивают значение Vd.
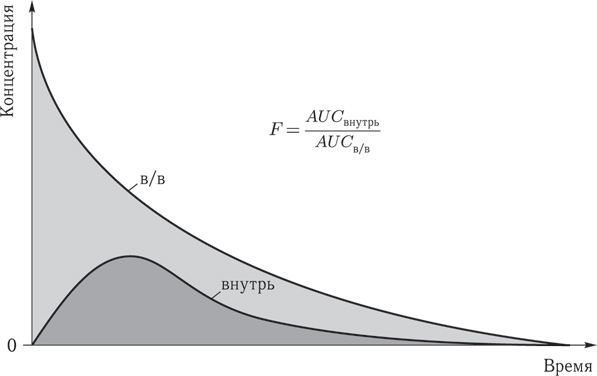
Биодоступность (f) – это показатель, описывающий поступление ЛС в системный кровоток при используемом пути введения по сравнению с внутривенным введением. Это часть введенного лекарственного вещества, достигшая системного кровотока относительно введенной дозы, выраженная в процентах. Препараты, вводимые внутривенно, имеют биодоступность 100 %.
Используя временной показатель, можно рассчитать площадь под кривой (AUC) на графике зависимости концентрации от времени. Отношение (AUC перорально /AUC в\в) x 100 позволяет определить биодоступность (f).
Рис. 2. Биодоступность (f) – это часть введенного лекарственного вещества, выраженная в процентах, достигшая системного кровотока относительно введенной дозы. (Пояснения в тексте.)
Метаболизм ЛС происходит подобно другим ксенобиотикам, попавшим в организм. ЛС метаболизируются во многих тканях, но преимущественно в печени, и этот процесс подразумевает превращение молекулы лекарства в легко выводимые метаболиты. Основная цель метаболизма лекарств – облегчение выведения чужеродных химических соединений за счет повышения их растворимости в воде (гидрофильности). Очевидно, что ферменты, метаболизирующие лекарства, развились как универсальная защита от чужеродных химических веществ, поступающих из окружающей среды.
Гидрофильные вещества легче выводятся с мочой, так как не подвергаются реабсорбции в почечных канальцах. При этом химические модификации в процессе метаболизма значительно чаще снижают, чем увеличивают фармакологическую активность ЛС. Метаболически активируемые, но исходно неактивные ЛС называются пролекарствами (например, активный метаболит антиагреганта клопидогрел образуется под воздействием фермента CYP2С19, входящего в систему микросомальных ферментов печени).
Некоторые ЛС в значительной степени или полностью инактивируются при первом прохождении через печень, в связи с чем их необходимо вводить сублингвально или парентерально. Так, биодоступность нитроглицерина при приеме внутрь составляет менее 1 % из-за эффекта первого прохождения через печень. Однако при сублингвальном приеме, при котором нитроглицерин всасывается, минуя воротную вену и печень, этот показатель возрастает до 38 %.
Основными эффекторами метаболизма лекарств являются ферменты цитохрома Р450 (CYP450) в печени.
Выделяют две основные фазы метаболизма. Большинство реакций фазы I осуществляют всего несколько монооксигеназ широкого спектра действия подсемейства CYP (цитохром Р450) 1–3, так называемых микросомальных ферментов печени. Наиболее часто в метаболизме ЛС участвует фермент CYP3A4. Фаза I метаболизма состоит из реакций восстановления, окисления или гидролиза. Реакции фазы I обычно превращают исходное лекарство в более полярный метаболит посредством образования групп – ОН, – NH2 или – SH.
Недостаточно полярные для элиминации препараты могут быть впоследствии (или первично) модифицированы ферментами фазы II, характеризующейся синтетическими реакциями. Наиболее частыми реакциями фазы II являются конъюгации с глюкуроновой кислотой. Кроме того, ЛС могут быть конъюгированы с глутатионом или глицином или модифицированы путем переноса метильных, ацетильных или сульфагрупп от соединений-доноров.
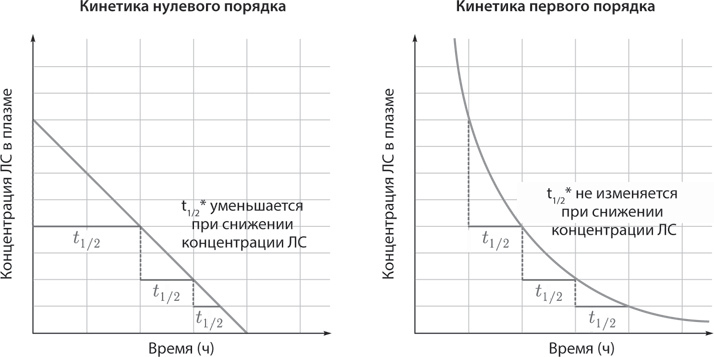
Выделяют два основных типа кинетики метаболизма лекарств. Кинетика нулевого порядка: скорость метаболизма и/или элиминации остается постоянной и не зависит от концентрации препарата в плазме в равновесном состоянии (Cплазма снижается линейно с течением времени).
Нулевой порядок – это метаболизм с ограниченной емкостью [этанол, фенитоин, аспирин (в высоких концентрациях)].
Кинетика первого порядка: скорость метаболизма и/или элиминации прямо пропорциональна концентрации препарата в плазме (Cплазма экспоненциально снижается с течением времени).
Первый порядок – это метаболизм, зависящий дозы (большинство ЛС; рис. 3).
Рис. 3. Типы кинетики метаболизма лекарственных средств. (Пояснения в тексте.) (* – период полувыведения, см. стр. 31)
Активность микросомальных ферментов печени может изменяться. Важной причиной снижения индивидуальной активности микросомальных ферментных систем являются инактивирующие мутации (полиморфизмы) генов, кодирующих микросомальные ферменты печени. Так, существуют аллели CYP2C19, кодирующие образование фермента со сниженной или отсутствующей функцией (обладатели таких полиморфизмов являются так называемыми слабыми метаболизаторами). У таких пациентов со сниженной активностью CYP2C19 при приеме пролекарства клопидогрела меньшее количество препарата переходит в активную форму, что проявляется его сниженной эффективностью. В результате возникает риск тромбоза коронарных сосудов на фоне формально правильно назначенных ЛС. Активность ферментов может снижаться на фоне препаратов, ингибирующих их активность (например, макролиды и коназолы), а также у пациентов с заболеваниями печени или недостаточным печеночным кровотоком. Метаболизм ЛС ускоряется у пациентов, получавших индукторы ферментов печени, метаболизирующих лекарственные препараты (фенобарбитал, зверобой продырявленный). Индукция и ингибирование метаболизма лекарств являются примерами фармакокинетических взаимодействий лекарств.
Скорость и интенсивность метаболизма ЛС варьирует не только среди пациентов, но даже у одного конкретного человека с течением времени (с этим связаны ограничения в дозах ЛП у детей и пациентов старше 60 лет).
Выведение, осуществляемое преимущественно почками, подразумевает удаление ЛС и его метаболитов из организма.
Почки являются основным (но не единственным) органом, выделяющим лекарство. Три компонента почечной экскреции: клубочковая фильтрация, секреция и реабсорбция – определяют скорость этого процесса. Лекарственные вещества, экскретируемые почками, попадают в мочу посредством клубочковой фильтрации. Кроме того, некоторые лекарственные средства выделяются в проксимальные канальцы переносчиками органических катионов и анионов. Липофильные препараты дополнительно секретируются в проксимальные канальцы посредством пассивной диффузии, так как они легко проникают через мембраны клеток нефрона.