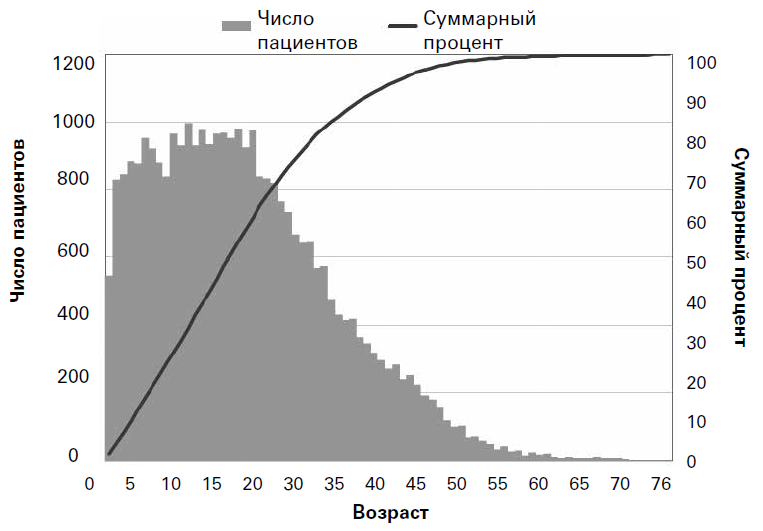
Касательно 2012 г. в регистре США сообщалось, что 49,1 % популяции составляли взрослые (18 лет и старше) при диапазоне возраста от рождения до 82 лет и медиане возраста, равной 17,7 лет (средняя – 19,8 лет)[7]. Регистр пациентов Европейского общества муковисцидоза (ECFSPR) является более новым, но в настоящее время содержит данные о более чем 32 000 пациентов из 22 европейских стран и охватывает широкий спектр социально-демографических и культурных характеристик[8]. В целом европейские данные очень сходны с данными из США: 49,3 % пациентов были классифицированы как взрослые (18 лет и старше) при широком диапазоне возраста от рождения до 80,1 лет и медиане возраста, равной 17,8 (средняя – 19,5) лет (рис. 2).
Рис. 2. Кросс-секционные данные о возрасте пациентов в Европе в 2010 г. (воспроизведено с разрешения Европейского общества муковисцидоза[9]) Каждый вертикальный столбик представляет число пациентов этого возраста в 2010 г. Суммарный процент (черная линия, правая вертикальная ось) показывает, сколько было пациентов (в процентах) младше определенного возраста (например, 50 % пациентов были младше 18 лет). В некоторых европейских странах доля взрослых пациентов особенно высока – как в Дании, где минимум 56 % больных MB являются взрослыми (18 лет и старше). Примерно 20 % популяции Европейского регистра составляют люди в возрасте старше 30 лет и 6 % – люди старше 40 лет. Однако существуют значимые вариации между регионами по социально-демографическим показателям, предположительно отражающие такие проблемы, как доступность медицинской помощи, поздняя диагностика, наличие средств и политика в области здравоохранения. Кросс-секционный анализ данных ECFSPR подчеркнул это, показав некоторые важные демографические различия между европейскими странами[10]. Медиана возраста пациентов из стран Европейского союза (ЕС) оказалась на 4,9 лет больше, чем в странах, не входящих в ЕС, и доля пациентов в возрасте 40 лет и старше также была более высокой в странах ЕС (5 % против 2 %). Важно то, что когда демографические показатели стран ЕС применили в моделях к странам, не являющимся членами ЕС, оказалось, что популяция пациентов с MB в этих странах может увеличиться на 84 %, что показывает поразительное потенциальное влияние, которое социально-демографические факторы могут оказывать на выживаемость. В Великобритании, согласно оценке, популяция взрослых пациентов с MB будет увеличиваться примерно на 145 пациентов в год[11]. Поскольку это соответствует числу больных в клинике для взрослых среднего размера, то имеет важные последствия не только для пациентов, но также и для обеспечения медицинской помощи и обучения персонала в будущем. 2. Почему пациенты, страдающие MB, теперь живут дольше? Причиной стабильного роста медианы продолжительности жизни с увеличением популяции взрослых является сочетание множества факторов. Они могут быть грубо классифицированы как генетические и негенетические факторы, которые включают условия окружающей среды (например, патогенные микроорганизмы, пол, воздействие табачного дыма и климат), факторы, связанные с системой оказания медицинской помощи (например, доступ к специализированным клиникам, меры по контролю инфекций и соблюдение пациентами рекомендаций врача), и социально-экономические факторы. Вследствие этого некоторые определяющие факторы выживаемости в большей мере поддаются модификации, чем другие; например, факторы, связанные с лечением, легче контролировать, чем факторы, связанные с климатом. С 1938 г. ключевые научные достижения, в том числе выявление повышенного содержания хлоридов в потовой жидкости в 1950 г. и открытие гена MB (регулятора трансмембранной проводимости при муковисцидозе (СFТR)) в 1989 г., являлись инструментами для улучшения нашего понимания патофизиологии MB[12][13][14]. Однако даже до этих событий имело место значительное улучшение оказания помощи в результате тщательного клинического наблюдения и системного подхода к лечению осложнений болезни (например, инфекции и недостаточности питания). Недостаточное питание, инфицирование патогенными микроорганизмами, такими как Pseudomonas aeruginosa и Burkholderia cenocepacia, отсутствие лечения диабета, связанного с MB, и женский пол являются точно установленными факторами, коррелирующими с более неблагоприятными исходами при MB[15][16][17][18]. Краеугольными камнями лечения в течение многих лет были внедрение интенсивных схем антибиотикотерапии, применение ферментов поджелудочной железы и оптимизация питания, эффективное очищение дыхательных путей, трансплантация легких, противовоспалительные препараты, препараты, усиливающие удаление мокроты (например, дорназа альфа), и, позже, внедрение специфических препаратов, воздействующих на CFTR при разных типах мутаций[19][20][21][22]. Эти факторы в сочетании с многоуровневым подходом к помощи при MB, обеспечиваемым мультидисциплинарной группой специалистов (MDT), играли главную роль в увеличении продолжительность жизни пациентов и послужили моделью для лечения других хронических заболеваний[23]. Когда был открыт ген CFTR, предположили, что некоторую вариабельность выживаемости при MB можно объяснить различиями мутаций; однако неоднократно было показано, что корреляция генотипа с фенотипом является слабой, за исключением состояния поджелудочной железы (например, достаточная функция или нарушенная)[24]. Мутации CFTR можно разделить на шесть классов в соответствии с их влиянием на функцию CFTR. Классы I–III обычно связаны с более тяжелым течением заболевания. В типичном случае у пациента, являющегося носителем одной из этих мутаций на каждой аллеле, MB диагностируется рано (т. е. в возрасте младше 1 года), и у него имеет место недостаточность поджелудочной железы[25][26]. Заболевание у пациентов с по меньшей мере одной мутацией классов IV–VI часто диагностируется позже, функция поджелудочной железы у них нередко сохранена и поэтому состояние питания лучше[27][28]. Некоторые мутации, связанные с сохранением функции CFTR, проявляются как нетяжелое поражение легких, но для огромного количества других мутаций это не так, и могут иметься различия между двумя субъектами с одним и тем же генотипом[29]. Предполагаемые механизмы для объяснения этого факта включают многочисленные влияния инфекций, питания и окружающей среды, хотя, вероятно, ни один из этих механизмов полностью не объясняет различия[30]. Мутации других генов (не CFTR) или полиморфизмы – обозначаемые термином «модификаторы генов» – по-видимому, вносят свой вклад в ситуацию, но до настоящего времени идентифицировано лишь ограниченное число модификаторов[31][32][33].
вернутьсяCystic Fibrosis Foundation Patient Registry. 2012 Annual Data Report. Bethesda, Maryland; 2013. Available from: www.cff.org/UploadedFiles/research/Clinical Research/PatientRegistryReport/2012-CFF-Patient-Registry.pdf вернутьсяZolin A, McKone EF, Van Rens J, et al. European Cystic Fibrosis Society Patient Registry Annual Data Report 2010. Karup: ECFS; 2014. Available from: https:// www.ecfs.eu/files/webfm/web-files/File/ecfs_registry/ECFSPR_ Report10_v12014_final_020617. pdf вернутьсяZolin A, McKone EF, Van Rens J, et al. European Cystic Fibrosis Society Patient Registry Annual Data Report 2010. Karup: ECFS; 2014. Available from: https:// www.ecfs.eu/files/webfm/web-files/File/ecfs_registry/ECFSPR_ Report10_v12014_final_020617. pdf вернутьсяMcCormick J, Mehta G, Olesen HV, et al. Comparative demographics of the European cystic fibrosis population: a cross-sectional database analysis. Lancet 2010; 375: 1007-13. вернутьсяDodge JA, Lewis PA, Stanton M, et al. Cystic fibrosis mortality and survival in the UK: 1947–2003. Eur Respir J 2007; 29: 522-6. вернутьсяdi Sant'Agnese PA, Darling RC, Perera GA, et al. Abnormal electrolyte composition of sweat in cystic fibrosis of the pancreas; clinical significance and relationship to the disease. Pediatrics 1953; 12:549-63. вернутьсяRiordan JR, RommensJM, Kerem B, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989; 245:1066-73. вернутьсяRommensJM, lannuzzi MC, Kerem B, et al. Identification of the cystic fibrosis gene: chromosome вернутьсяCorey M, Farewell V. Determinants of mortality from cystic fibrosis in Canada, 1970–1989. Am J Epidemiol 1996; 143: 1007-17. вернутьсяIsles A, Maclusky I, Corey M, et al. Pseudomonas cepacia infection in cystic fibrosis: an emerging problem. J Pediatr 1984; 104: 206-10. вернутьсяMilla CE, Billings J, Moran A. Diabetes is associated with dramatically decreased survival in female but not male subjects with cystic fibrosis. Diabetes Care 2005; 28: 2141-4. вернутьсяChotirmall SH, Smith SG, Gunaratnam C, et al. Effect of estrogen on pseudomonas mucoidy and exacerbations in cystic fibrosis. N Engl J Med 2012; 366:1978-86. вернутьсяRamsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 2011; 365: 1663-72. вернутьсяSaiman L, Marshall ВС, Mayer-Hamblett N, et al. Azithromycin in patients with cystic fibrosis chronically infected with Pseudomonas aeruginosa: a randomized controlled trial. JAMA 2003; 290: 1749-56. вернутьсяFuchs FIJ, Borowitz DS, Christiansen DH, et al. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N Engl J Med 1994; 331: 637-42. вернутьсяPryor J, Tannenbaum E, Scott S, et al. Beyond postural drainage and percussion: airway clearance in people with cystic fibrosis. J Cyst Fibros 2010; 9: 187-92. вернутьсяMahadeva R, Webb K, Westerbeek RC, et al. Clinical outcome in relation to care in centres specialising in cystic fibrosis: cross sectional study. BMJ 1998; 316: 1771-5. вернутьсяHamosh A, Rosenstein B, Nash E, et al. Correlation between genotype and phenotype in patients with cystic fibrosis. N Engl J Med 1993; 329: 1308-13. вернутьсяMcKone EF, Emerson SS, Edwards KL, et al. Effect of genotype on phenotype and mortality in cystic fibrosis: a retrospective cohort study. Lancet 2003; 361: 1671-6. вернутьсяMcCloskey M, Redmond A, Hill A, et al. Clinical features associated with a delayed diagnosis of cystic fibrosis. Respiration 2000; 67: 402-7. вернутьсяNickJA, Rodman DM. Manifestations of cystic fibrosis diagnosed in adulthood. Curr Opin Pulm Med 2005; 11:513-18. вернутьсяAugarten A, Yahav Y, Szeinberg A, et al. Mild cystic fibrosis and normal or borderline sweat test in patients with the 3849+ 10 kb C→T mutation. Lancet 1993; 342: 25–6. вернутьсяMcKone EF, Emerson SS, Edwards KL, et al. Effect of genotype on phenotype and mortality in cystic fibrosis: a retrospective cohort study. Lancet 2003; 361: 1671-6. вернутьсяSchechter MS. Non-genetic influences on cystic fibrosis lung disease: the role of sociodemographic characteristics, environmental exposures, and healthcare interventions. Semin Respir Crit Care Med 2003; 24: 639-52. вернутьсяBlackmanS, HsuS, RitterS, etal. A susceptibility gene for type 2 diabetes confers substantial risk for diabetes complicating cystic fibrosis. Diabetologia 2009; 52: 1858-65. вернутьсяBartlett JR, Friedman KJ, Ling SC, et al. Genetic modifiers of liver disease in cystic fibrosis. JAMA 2009; 302: 1076-83. вернутьсяDrumm ML, Konstan MW, Schluchter MD, et al. Genetic modifiers of lung disease in cystic fibrosis. N Engl J Med 2005; 353: 1443-53. |