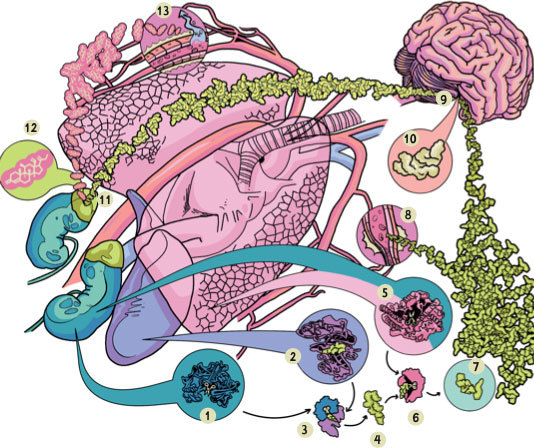
Рисунок 7. Схема работы ренин-ангиотензин-альдостероновой системы:
1 – секреция ренина почками в ответ на снижение внутрипочечного давления; 2 – секреция ангиотензиногена печенью; 3 – отщепление ренином декапептида от ангиотензиногена, высвобождение ангиотензина I; 4 – ангиотензин I; 5 – синтез ангиотензинпревращающего фермента (АПФ) в легких и почках; 6 – преобразование ангиотензина I в ангиотензин II под воздействием АПФ, который отщепляет от него две последние аминокислоты; 7 – ангиотензин II; 8 – сосудосуживающая активность ангиотензина II, за счет которой растет центральное давление; 9 – стимуляция гипофиза ангиотензином II, в результате чего происходит секреция антидиуретического гормона (АДГ); 10 – АДГ; 11 – стимуляция АДГ выброса альдостерона надпочечниками; 12 – альдостерон. 13 – альдостерон задерживает воду и повышает проницаемость мембран клеток, за счет чего увеличивается объем циркулирующей крови и растет кровяное давление
Избыточный синтез одного из участников цепочки РААС в печени (ангиотензиногена), к примеру, приводит к повышенной жесткости тканей сердца – в результате чрезмерного синтеза белков матрикса фибробластами. В итоге работа сердца сильно страдает73, 74, 75, 76, 77. Это происходит следующим образом: активация рецептора к ангиотензину II AT1, который представлен на поверхности клеток многих тканей (сердце, почки, нервная система и др.), вызывает избыточные ответы в кардиомиоцитах и синтез белка внеклеточного матрикса фибробластами сердца.
В фибробластах сердца ангиотензин II повышает синтез коллагена, фибронектина, ламинина и остеопонтина. Гладкомышечные клетки сосудов, стимулированные ангиотензином II, демонстрируют увеличение количества мРНК коллагена, фибронектина, ламинина и тенасцина.
Решение вышеописанных проблем уже проглядывается: опыты на животных показали, что фармакологическое ингибирование компонентов РААС приостанавливает разрастание компонентов матрикса, приводящее к фиброзу, а значит, и патологические процессы в тканях сердца78.
В почках ангиотензин II стимулирует синтез коллагенов, фибронектина и ламинина мезангиальными клетками78, 79.
Способность ангиотензина II стимулировать продукцию TGF-β[14], одного из главных профиброзных факторов, связывают с развитием возрастной сосудистой гипертрофии.
Постоянная активация рецепторов TGF-β приводит к аномальному накоплению соединительной ткани в почках и сосудах, ведущему к фиброзным патологиям80, 81, 82.
Кроме того, продукция TGF-β может индуцироваться при увеличении количества рецепторов RAGE[15]83. Повышение экспрессии этого цитокина может быть прямым следствием процессов гликирования. Возможно, если получится изобрести препарат, «отменяющий» гликирование, вред от избытка TGF-β тоже снизится84, 85, 86.
Таким образом, жесткость почки (за счет синтеза коллагена) растет как напрямую от воздействия ангиотензина II, так и в результате продукции TGF-β. Также запускается очередной порочный круг (на этот раз не только в почках, но и в сосудах): от роста количества КПГ (которые для матрикса и есть сшивки) растет уровень TGF-β, который еще сильнее ускоряет разрастание соединительной ткани.
Митохондрии и матрикс – продолжение истории
Митохондрии находятся внутри клетки, матрикс – снаружи. Поэтому работа митохондрий и матрикса тесно связана друг с другом посредством цитоскелета.
Для обновления белков основных структур цитоскелета (микрофиламенты, промежуточные филаменты, микротрубочки) требуется энергия аденозинтрифосфата (АТФ). Поэтому митохондрии в клетке постоянно двигаются, собираясь в местах, где высока потребность в АТФ94. В свою очередь, правильная организация скелета клетки важна для нормального функционирования митохондрий – они тесно взаимодействуют с ним, чтобы поддерживать свою морфологию. Ангиотензин II, о котором мы говорили выше, нарушает нормальную организацию цитоскелетных филаментов, что негативно сказывается на работе митохондрий94.
Как же взаимосвязаны изменение матрикса, нарушение работы цитоскелета и митохондрии?
Ангиотензин II вызывает изменения в процессе синтеза компонентов межклеточного матрикса, что влияет и на цитоскелет клетки. Известно, что если эндотелиальные клетки окружены более жестким матриксом, то их микротрубочки образуются дольше, правда, получаются в итоге более прочными. Клетки, находящиеся на менее жестком матриксе, образуют не такие прочные микротрубочки, но и растут они быстрее95. Микротрубочки, в свою очередь, очень важны для функционирования митохондрий: при их «разборке» митохондрии теряют свою подвижность. Взаимодействие митохондрий с более прочными трубочками ухудшает их структуру и нарушает энергетическую систему в клетке96.
Еще один из механизмов нарушения цитоскелета клетки, помимо действия ангиотензина II, – регуляция уже упомянутым цитокином TGF-β[16] реорганизации актиновых филаментов. Вызывая перегруппировки актиновых филаментов, он влияет на рост и дифференцировку клеток, так как в ядре запускается действие определенных транскрипционных факторов84, 85, 86 (рис. 8).
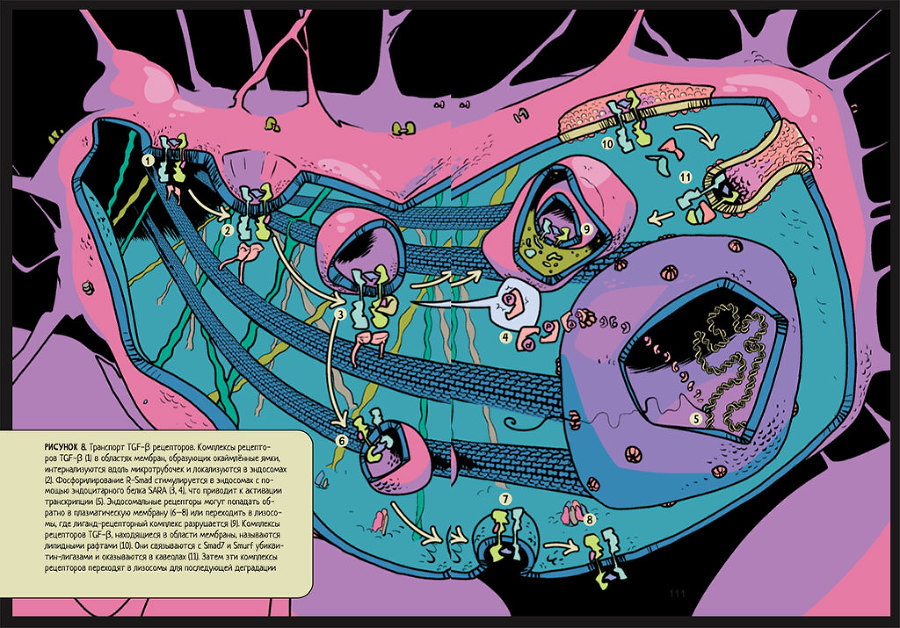
Рисунок 8. Транспорт TGF-β рецепторов. Комплексы рецепторов TGF-β (1) в областях мембран, образующих окаймленные ямки, интернализуются вдоль микротрубочек и локализуются в эндосомах (2). Фосфорилирование R-Smad стимулируется в эндосомах с помощью эндоцитарного белка SARA (3, 4), что приводит к активации транскрипции (5). Эндосомальные рецепторы могут попадать обратно в плазматическую мембрану (6–8) или переходить в лизосомы, где лиганд-рецепторный комплекс разрушается (9). Комплексы рецепторов TGF-β, находящиеся в области мембраны, называются липидными рафтами (10). Они связываются с Smad7 и Smurf убиквитин-лигазами и оказываются в кавеолах (11). Затем эти комплексы рецепторов переходят в лизосомы для последующей деградации.
Аиша Мелуан и ее коллеги показали, что белок SPARC[17] влияет одновременно и на изменения состава внеклеточного матрикса, и на функцию митохондрий в мышечных клетках100.
В мышцах SPARC синтезируется при строительстве или заживлении мышечной ткани. Еще он обладает способностью связываться с коллагенами разных типов, за счет чего влияет на перестройку и формирование внеклеточного матрикса102.
Что касается митохондрий, то этот белок влияет на их развитие путем взаимодействия с индуктором биогенеза митохондрий – белком AMPK (Adenosine Monophosphate-activated Protein Kinase, «протеинкиназой, активируемой аденозинмонофосфатом»)103. Таким образом, SPARC, как и ангиотензин II, и TGF-β, может выступать связующим звеном между работой митохондрий и процессами, протекающими в межклеточном матриксе.
Та же группа ученых представила еще один механизм взаимодействия митохондрий с матриксом при помощи SPARC104. Схема их непростых взаимодействий представлена на рисунке 9.
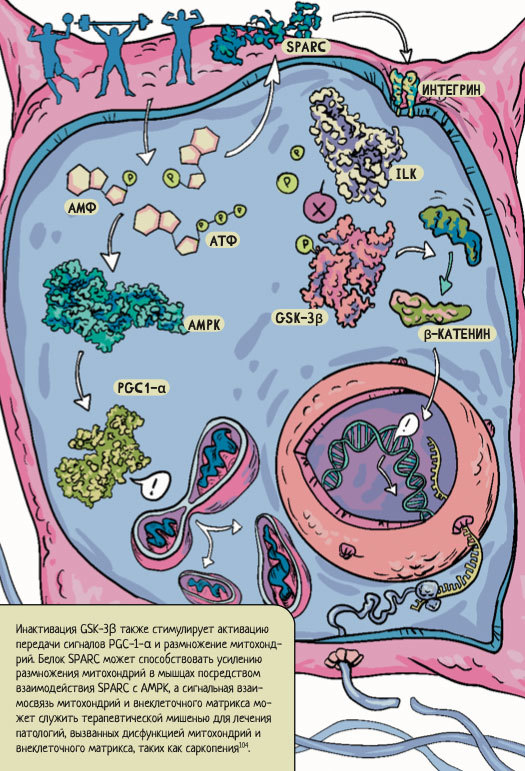
Рисунок 9. Влияние белка SPARC на экспрессию внеклеточного матрикса и на репликацию и транскрипцию митохондриальной ДНК. Физические упражнения вызывают активацию AMPK за счет увеличения соотношения аденозинмонофосфата к аденозинтрифосфату (AMФ/ATФ). Это, в свою очередь, индуцирует биогенез митохондрий посредством активации гамма-рецептора, активируемого пролифератором пероксисом коактиватора-1-альфа (PGC1-α), и повышает уровень экспрессии белка SPARC, что активирует интегрин-связанную киназу (ILK). Последняя фосфорилирует и инактивирует киназу гликогенсинтазы-3-бета (GSK-3ß), что приводит к стабилизации β-катенина и выработке белков мышечного внеклеточного матрикса. Инактивация GSK-3β также стимулирует активацию передачи сигналов PGC-1-α и размножение митохондрий. Белок SPARC может способствовать усилению размножения митохондрий в мышцах посредством взаимодействия SPARC с AMPK, а сигнальная взаимосвязь митохондрий и внеклеточного матрикса может служить терапевтической мишенью для лечения патологий, вызванных дисфункцией митохондрий и внеклеточного матрикса, таких как саркопения104.