ОЖИРЕНИЕ И ДИАБЕТ: РАЗНЫЕ МУТАЦИИ В РАЗНЫХ ГЕНАХ
Вспомнив, в чем состояла важность открытий Менделя, вернемся к нашим мышкам в лаборатории Джексона. Ученые уже знали, что болезнь мышей была результатом мутации одного-единственного гена. Ожирение у них было связано с бесплодием, в то время как их родители не были ни толстыми, ни бесплодными. Это означало, что каждый из родителей нес в себе одну дефектную хромосому (как и показывает скрещивание 2 в эксперименте Менделя), и только если их отпрыски наследовали две хромосомы (как в случае с зеленым горошком), по одной от каждого родителя, они становились толстыми и бесплодными. Это, как мы видим, случай рецессивной наследственности.
Если мыши с диабетом и ожирением были похожи почти во всем, означало ли это, что мутация произошла в одном и том же гене? Нужно иметь в виду, что этот эксперимент проводился, когда у генетиков еще не было современной техники и кроме того, что это мутация одного рецессивного гена, они ничего больше не знали. Чтобы ответить на этот вопрос, Даг Коулмен был вынужден начать серию экспериментов со своеобразным «парабиозом». В ходе этих экспериментов две мыши были пришиты одна к другой, деля одну кровеносную систему, – из них сделали своего рода сиамских близнецов. Зрелище не из приятных, и, сразу скажем, сейчас такие эксперименты уже не проводятся. Но тогда было другое время.
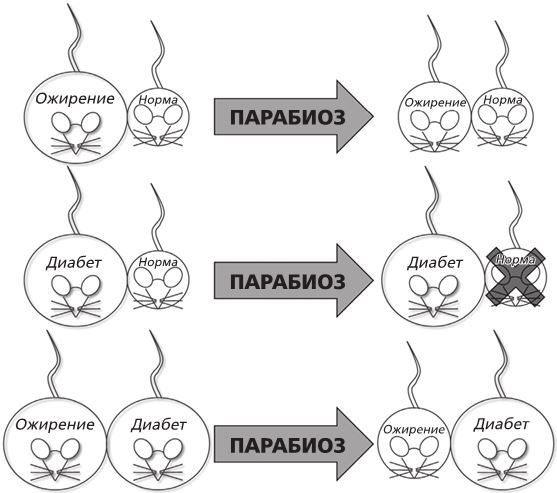
Рисунок 4. Парабиотические эксперименты Дага Коулмена, которые привели к появлению гипотезы о гормоне сытости.
Провели три эксперимента. В первом ученый соединил мышь с ожирением и нормальную мышь. Он увидел, что с нормальной мышью ничего не случилось, а толстая сильно похудела. Во втором он соединил мышь, больную диабетом, и нормальную. На этот раз нормальная мышь перестала есть, потеряла вес и в конце концов умерла, а с больной ничего не случилось: она продолжала есть и не теряла веса. В третьем эксперименте «сиамскими близнецами» оказались мышь с ожирением и мышь с диабетом. Как и в первом случае, мышь с ожирением перестала есть и стала терять вес, а мышь с диабетом вела себя так же, как и до эксперимента[12]. Все это было очень странно.
Тем не менее самой очевидной частью эксперимента стало то, что мышь с ожирением и мышь с диабетом страдали от мутаций в разных генах, потому что они вели себя по-разному, когда их соединяли либо с мышью нормального размера, либо друг с другом. Не такой очевидной (по крайней мере, для меня) оказалась идея, которую высказал Даг. Он предположил, что у мыши с ожирением не хватало «маркера сытости» – назовем его так, – это вещество находится в кровеносной системе, и когда его количество растет, мышь прекращает есть. У мыши с диабетом проблем с «маркером сытости» не было, только она не могла на него реагировать. Удивительное заключение, конечно. Но вот какая тут была логика: когда мышь с ожирением была присоединена к нормальной мыши, она неожиданно стала получать сигнал о том, что наелась, потому что этот сигнал присутствовал в кровообращении нормальной мыши. В результате она прекратила есть слишком много и стала терять вес. Во втором эксперименте мышь с диабетом была к этому фактору нечувствительна: ее тело производило огромные количества этого вещества, так что когда нормальная мышь получала его вместе с кровью, она немедленно прекращала есть. Третий эксперимент, когда были соединены мыши с диабетом и ожирением, доказал, что мутировавшие гены двух мышей взаимодействовали друг с другом, потому что высокий показатель насыщения у мыши с диабетом передался мыши с ожирением, и тогда она прекратила есть.
Такова, по крайней мере, была гипотеза. И теперь надо было проверить еще одну маленькую деталь.
ЛЕПТИН – ГЕН ГОЛОДА?
Только в 1980-х годах бурное техническое развитие в области генетики позволило команде из Рокфеллеровского университета в Нью-Йорке начать исследование, которое действительно позволило продвинуться вперед в проверке этой гипотезы. В 1994 году, через 45 лет после того, как в одной из лабораторных клеток была замечена мышь с ожирением, лаборатория Джеффри Фридмана сообщила, что у таких мышей происходит мутация в гене, который Джефф решил назвать лептином[13].
Лептин, как выяснилось, производится в жире и затем поступает в кровь, где циркулирует как гормон. Когда Джефф получил лептин и впрыснул его мышке с ожирением, она перестала есть и похудела, подтвердив таким образом, что раньше у нее был недостаток нормального лептина. Это и был таинственный «гормон сытости», о котором говорил Даг. Еще через пару лет у мыши с диабетом была найдена мутация в рецепторе лептина, потому-то она и не худела, когда ей давали лептин[14]. И опять-таки предположение Дага – о том, что в крови у этой мыши был лептин, но она не могла его правильно воспринимать, – оказалось поразительно точным.
То, что ученые обнаружили лептин и его рецептор, впервые указало на присутствие в организме такой гормональной системы, которая может регулировать прием пищи и ничего не имеет общего с силой воли. Но затем появился еще один важный вопрос. Присутствует ли эта странная система только у мышей или же ею обладают и другие млекопитающие? Чтобы ответить на этот вопрос, в начале 1997 года к исследованиям подключились мой старый коллега Стивен О’Райли и его (в то время) студентка Садаф Фаруки. Как мы помним, приводившая к ожирению мутация в гене лептина не была создана искусственно, она получилась сама собой. Стивен – усатый ирландец, который любит поразмышлять. И в ходе своих размышлений он пришел к такому предположению: если мутация в гене могла привести к ожирению у млекопитающего, вероятно, подобные случаи ожирения есть и у людей. В одной из лабораторных морозильных камер у Стивена имелись пробирки с образцами крови двоих детей – они были двоюродными братом и сестрой, оба страдали ожирением. Садаф попыталась измерить лептин в этих образцах, но не смогла. Вернее сказать, анализ не показал присутствия лептина. Тогда Карл Монтагю, который проходил у Стивена постдокторантуру, определил строение гена лептина у этих детей и выявил мутацию – отсутствие одного-единственного гуанина (помните четыре элемента, из которых собрана ДНК: аденин (А) тимин (Т), гуанин (Г) и цитозин (Ц)). Нехватка гуанина была причиной того, что у детей в организме отсутствовал правильно функционирующий лептиновый белок. Стив в очередной раз оказался прав. Его команда в Кембриджском университете обнаружила – впервые в истории генетики, – что мутация в одном гене может привести к тяжелым формам ожирения у людей[15].
Как выглядели эти дети, которые не могли производить лептин? Младшему из них – назовем его Джон Б. – было три года. Он весил 42 килограмма. Я, например, вешу 75 килограммов. Вес Джона Б. – это 2/3 моего веса. Ожирение случилось не из-за того, что он слишком долго играл в видеоигры, пил слишком много сладких напитков и тому подобное – здесь налицо был случай серьезной болезни. Джон Б. родился с нормальным весом, но когда после грудного молока он перешел на твердую пищу, случилось что-то странное. Он постоянно был очень-очень голоден. Он страдал гиперфагией (от греч. hyper – «чрезмерно» и phagein – «поедать»). Это ненормальное пищевое поведение. То есть, к примеру, вы не можете сказать: «Слушай, на меня в выходные нашла такая гиперфагия!» или «Мы поехали в Грецию, и там у меня случился приступ гиперфагии». Нет, у вас все хорошо, вы просто переели. Гиперфагия означает, что родителям этих детей пришлось купить навесной замок на морозильник, иначе дети сами открывали морозилку и ели замороженные рыбные палочки. Неудивительно, что содержание жира в организме у девочки и мальчика было невероятно высоким: около 57 % (у нормального человека в зависимости от пола оно составляет 15–28 %). Неожиданным было то, что старшая девочка – назовем ее Джейн А. – не выказывала никаких признаков полового созревания (мальчик был в то время слишком мал, чтобы о чем-либо говорить), и у обоих неважно работала иммунная система. На первый взгляд, странная комбинация характеристик: ожирение, необычное пищевое поведение, бесплодие и расшатанная иммунная система. Но я вернусь к этому позднее и покажу, что ничего странного в этом нет – наоборот, все логично… даже поедание замороженных рыбных палочек.